
Reforming Psychological Research: Lessons from the Success of Psychiatric Genetics
Reforming Psychological Research: Lessons from the Success of Psychiatric Genetics
Field wide collaboration, a firm grip on file drawers, false positives, HARKing, and multiple testing control, combined with a rigorous theoretical foundation, formed a recipe for success.

It may be counterintuitive for some to hear psychiatric genetics referred to as a success. Its been a decade and a half since the first genome wide association studies (GWAS) of psychiatric outcomes, initially schizophrenia12 where published, and almost a decade since the first resoundingly successful GWAS of schizophrenia3. While since then a lot (a LOT) of genetic variants, both common and rare4 , have been identified for outcomes like schizophrenia diagnosis, these have not yet resulted in novel therapies, clinically useful prediction of outcome or clinical course. Arguably these GWAS have facilitated slightly better biological understanding of psychiatric phenomena, but that is a contested conclusion.
While I personally think judging the yield of psychiatric genetics now is shortsighted and inpatient, that not the point of this post. In this post I want to consider the success of psychiatric genetics as a research field, and it would be a terrible idea to asses the quality of the research process based on substantive results. Just like we can’t conflate the quality of a randomised trial with whether it confirms the treatment works, we can’t judge psychiatric genetics as a field on the utility or immediate translation its findings. Rather I think we should evaluate the process by which theory formation, data collection, and evaluation of theories against data, inform research in the field, and lead to concrete outcomes (both rejection and confirmation of prior believes) that enable progress.
An abbreviated description of the overall goals of psychiatric genetics in the early 2000’s might have described two key steps. First, to establish which genetic variants relate to psychiatric outcomes of interest and what the magnitude of their effect was. At the time psychiatric genetics became feasible there was ongoing debate on how many gentic variants would relate to diagnosis and what that might mean for the clinical utility of the findings, and the time and effort needed to identify all variants of relevance. A second goal for the field that was widely shared, was to then identify the neuro-biological or epidemiological5 (though the neuro-biological goals seem to be mentioned more frequently in the literature at the time) mechanisms via which genetic variants influence psychiatric outcomes. In this post I consider success in psychiatric genetics as success in terms of the first key step, I will at some point write about why I feel the second key step has remained elusive.
In this post I’ll argue psychiatric genetics appears to have had three things going for it. First, the field has had solid governmental and philanthropic funding. This mattered greatly for its success, but it is not entirely unique to the field. The sequencing of the human genome obviously captured the imagination and generated goodwill as well as governmental and philanthropic investment. Investment also spilled over into psychiatric genetics.

Second, psychiatric genetics benefitted from tight type-1 error control and an social/analytical process that prevented HARKing, fishing, and/or noise mining. The process of meta-analysis in the field resembles a loose mix of pre-registration and registered reporting. While these procedures were informal and imperfect compared to formal registration, they did prevented widespread HARKing, file-drawer problems and research waste. Third, specifically the genetics part in psychiatric genetics has a strong theoretical foundation that guides study design, accurate and cheap measurement of the genome, efficient data collection, contributes to tight type-1 error control, and enables the forecast of expected results at future sample sizes.
As with any retrospective reflection this post has a strong risk of being influenced by hindsight bias. I risk inferring seemingly intentional deliberate progress from selective highlights that merely appear retrospectively correct, coherent or valuable. Its safest to view this piece as descriptive, personal and coloured by my experiences, and (VERY) amateur history. I wasn’t around for most of this, I wouldn’t be able to judge whether all of this was intentional and deliberate or only appears that way trough my lens. Please let me know what I got wrong, if people do I’ll discuss the criticism and feedback in a future post, I am mostly just very interested in getting this right. Finally, I discuss what I feel holds back further success in psychiatric genetics, which might be the lack of firm theoretical grounding for psychiatry, psychology and biological psychiatry when compared to genetics.
Tight control of multiple testing
A genome wide association study (GWAS), one of the key methods to link the genome to an outcome (“phenotype”), involves anywhere from 200.000 (early days) to 10.000.000 (current standard) statistical tests, each the association between the outcome and genetic variant. In a GWAS the alpha (p-value cut-off) clearly isn’t 0.05, but it also doesn’t depend on the number of associations tested. Because of the biological realities of how you inherit your DNA we inherit long consecutive segments from either parent combined with our natural history as a species this results in a genome where variants physically near eachother on the chromosome correlate strongly. Based on theory, models build on that theory, and early datasets, people where able to model the approximate number of independent “tests” or variants in the genome. The number of independent tests was estimated at ±1.000.000 and so the alpha or p-value cutoff for a GWAS was put at 5*10^-8. It doesn’t matter how many variants you prune or toss out for quality control reasons, the (p-value) bar doesn’t move. In some cases, if authors present work in which they restrict their search to a theory informed subset of markers (“only genes in neurotransmitter systems”) reviewers and readers alike will either insist on the genome wide significance threshold, or just evaluate agains that standard while reading anyway. The is no credible way of lowering the bar, which restricts researcher degree of freedom and reduces the risk of intentional or unintentional noise mining, HARKing etc.
However, as psychologists and other social scientists know, only part of the research degrees of freedom arise prior to doing an analysis or involve explicitly influencing the testing burden. Having a strict and theoretically informed threshold, doesn’t prevent a researcher from for example reconsidering their phenotype definition, or covariates if the GWAS yields an unexpected (or in the views of the overeager researcher undesirable) null result. Wouldn’t people, as happens in other fields, just run a few variations of a GWAS presenting the “optimal” result or dig deeper into technical issues when they find a null?
Research culture ensured pre-registration like procedures for GWAS
It seems the social norms/customs in the field that prevented selective re-analysis, fishing and HARKing arose from two events. First, there were initial GWAS studies that where underpowered and unsuccessful the first GWAS of schizophrenia had ±150 cases and controls, the second ±700 (see footnotes 1 and 2). Secondly, this failure to find common genetic variants with large effects on outcomes like schizophrenia, was one of the expected outcomes based on genetic theory. Some in psychiatry had suggested as far back as the 60’s that psychiatric outcomes like schizophrenia were most likely inherited not trough a single risk allele but trough many with modest effects6. If this was true, sample size requirement for GWAS would be huge, especially considering a GWAS involves a million tests. The basic power requirements requirements of GWAS where outlined based on theory driven power calculations in the 90’s, years prior to the first GWAS, when they still published a power calculation in Science7. While their analysis was optimistic in terms of effect sizes wrt psychiatric outcomes, it was visionary to outline based on genetic theory, how by sampling just the right parts of the genome, at a scale unherd off at the time (many thousands), would pinpoint genomic regions related to human disease.
When early psychiatricGWAS failed to identify a handful of common genetic variants with large effects on psychopathologies, it didn’t lead to the rejection of GWAS. They didn’t have to because there was firm theoretical reasons to keep going. Given those pre-existing theoretical considerations the solution was obvious: increase sample size, and pool results. When further early analyses of pooled data didn’t yield much in terms of genome wide significant results by 2009, they could leverage theory to design a prediction experiment that would tell them they were on the right track and that schizophrenia was in fact influenced by incredibly many genetic variants each with modest, small or very small effect. The experiment used polygenic scores and I have a separate post detailing that paper and some of its nuances:
Coordinated team sciences provided the social framework for better science
Pooling results across studies and groups on a genome wide scale required intense coordination to make sure analyses where comparable. Groups used different measurements of the genome, each of which covered a different set of variants. However, based on theory and empirical observation of the very high corrections between neighbouring genetic variants (a correlation that decays with distance, and the behaviour of which can be modelled based on theory8) the genotypes as measured by different research groups, could accurately be imputed to a common intersecting set of variants. Genetic theory helped solve the problem of finding a common measure of the genome, ensuring as far as genetics was concerned the data can be integrated across groups. This immutiation isnt to be confused with findign a common map for different MRI experiments whereh the common map is some ephemeral average map of conviencience, rather we are imputing the underlying genomic reality to a very ghigh degree of accuracy (yet far from perfect!).
In a meta-analysis each group would perform an association analysis according to a common protocol, and share results to a central analysis team for meta-analysis. Note that unlike a usual meta-analysis in psychology this procedure meant groups: 1. Agreed to participate before analyses were performed, 2. Agreed to a whole set of analytical and measurement parameters and procedures before results were known 3. Coordinated on analysis and results ensuring multiple pairs of eyes on all or most pieces of the pipeline/analysis. Furthermore, GWAS where computationally expensive, so both social pressure, and feasibility would have prevented frequent re-analysis for anything but very obvious technical error, strongly inhibiting fishing excursions. The procedure also prevented results from specific groups being dropped from the analysis for any but very apparent technical errors, as no group would risk being left out of the final analysis and paper. The process closely resembles the “many-labs” type studies in psychology where many groups agree to perform the same experiment(s) to ensure power, generalisability, fault tolerance and replicability.
The scientific culture and process of GWAS meta-analysis was invented in parallel by numerous consortia of research groups across complex trait genetics. Because many research groups contributed data and analysis to multiple consortia, best practices spread quickly. The research culture that arose, implemented all, or most, of the guardrails a study pre-registration would offer. I wouldn’t speak of this era and research culture so confidently, if its results hadn’t been stress tested much like various coordinated replication projects stress tested replicability of results across psychological and other social sciences. Psychological sciences went trough an (rude) awakening when systematic replication failed for 40 out of 100 studies sampled for a large replication project9. While individual consortia in genetics made a point of reproducing their previous findings in new data (sucessfully) the real field wide evaluation arose, again, from genetic theory.
Evaluation whether type-1 error is/was in fact well controlled in GWAS.
The expected effect for the effect of a genetic variant on an outcome, is its true effect size, plus bias, plus noise. I promise I'll get to the bias and noise in a future post. Due to the pervasive correlation between adjacent genetic variants across the genome, and the fact that variants are considered marginally (one by one, with no other variants in the model), the estimated effect size of any give variant also depends on the degree of correlation with its neighbours (some of whom would have a true causal effect). This was a known phenomena, but the degree to which it influences GWAS results wasn’t widely appreciated. In 2015 a group showed a measure of the total correlation between any variant and all its neighbours would correlate with true signal in a GWAS, while certain biases, noise, or falsification of results, would not10. The technique retroactively seperated noise, bias due to population stratification and other sources, and true signal. Its worth noting it would have also detected systematic falsification of results, unless the falsification would have been sophisticated enough to presage this relation between the genomic structure and true signal. Evaluation of all or most GWAS large enough for consideration on the whole resulted in a very favourable signal to noise ratio, suggesting widespread result falsification, pervasive biases or accidental or intentional noise mining played no or no appreciable result in GWAS.
For a more visceral snapshot of difference in replication success, consider a direct contrast (Figure 2 below) between the original and replication effect sizes in psychological sciences across 100 studies, and the original and replication of 1503 genetic variants across two independent GWAS of the same educational outcomes.
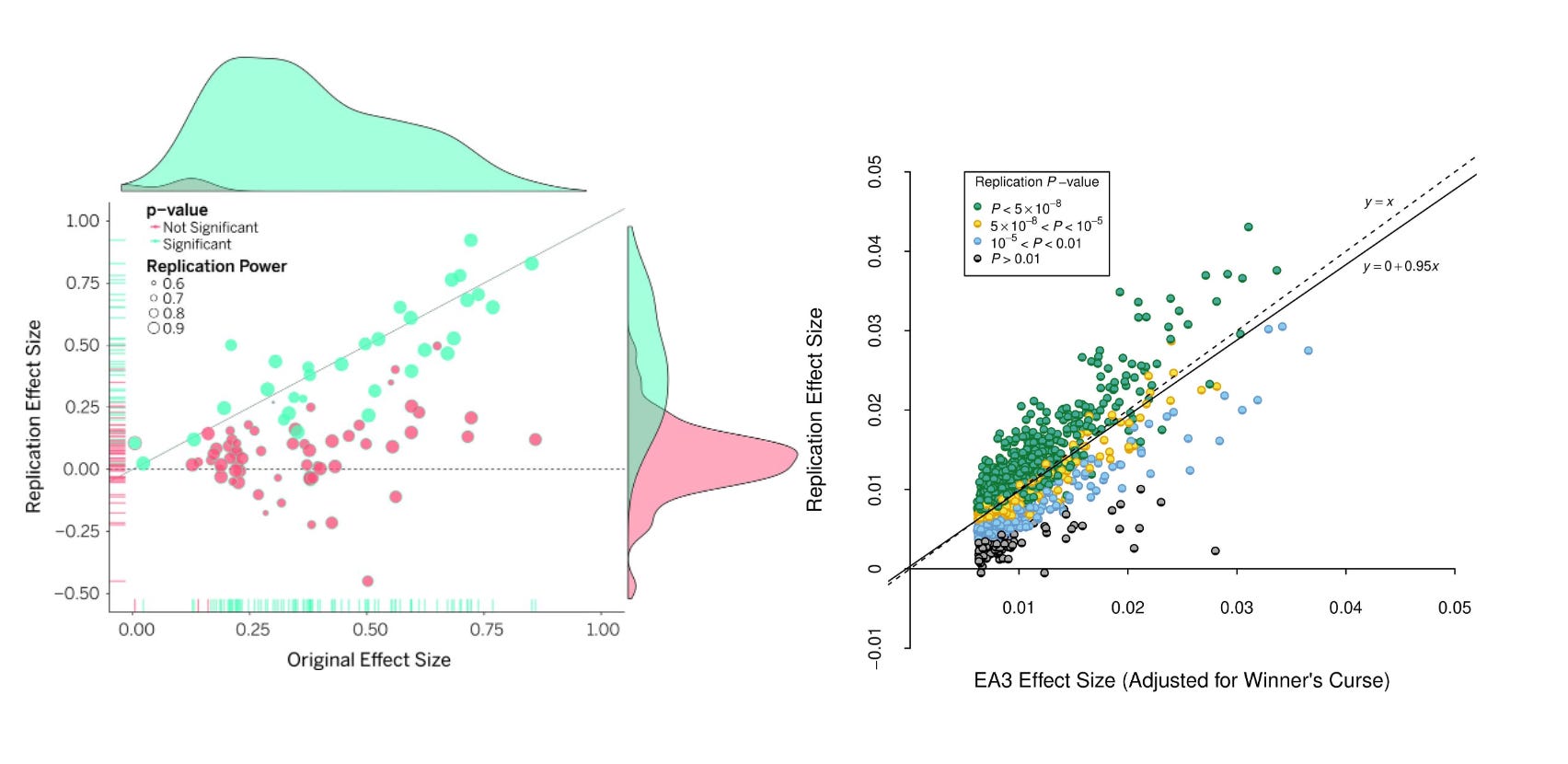
Note how the GWAS has very solid replication rates, where psychological studies do not? One caveat is that both GWAS where run by the same consortium, a team that would have conflicts of interest in showing their prior analysis had been solid. However, there are examples of two independent groups running a GWAS of the same trait on open data, near perfectly reproducing each others results11.
Sound genetic theory guided, or corrected, decision making
Early pioneers of genetic theory, a number of whom held eugenic and deeply disturbing ideas and believes we should recon with as a field, mapped out how genetic variants where likely inherited, how evolution might constrain certain variants with a deleterious effect, how a trait, or risk for a trait could be continuously distributed while variants where not. These rudimentary theories where strengthened and revised as the structure and exact mode of inheritance of the human genome was discovered. As early as the 60’s psychiatric researchers could theorise that the way which schizophrenia was inherited in families was incompatible with simple mendelian inheritance where a single or a few additive or dominant variants, but rather possibly meant schizophrenia is caused by many variants. These polygenic models, combined with theoretical analysis of what a genome wide association studies would involve before the human genome was mapped fully, meant that as the results of the first analysis cdid’t land in a theoretical vacuum. As results rolled in, the next steps to take as a field where clear or at least could be theorised, modelled and or reasoned about.
When early GWAS results where seemingly disappointing in terms of the number of loci identified, statistical analysis grounded in genetic theory12 pinpointed this as a consequence of very high polygenicity (±10% of independent regions in the genome may harbor a variant with some non-zero effect on schizophrenia) and not some technical failure in gonotyping, meta-analysis, process error or measurement issue. Similarly, as highlighted on twitter the other day, a 2014 paper13 on how to design association studies when vriants are very rare (a type of variant missed by GWAS), is so well grounded in theory that it forms a roadmap for the analysis of rare variants which is more or less a complete description of how rare variant analysis of spychiatric disorders is conducted to this day.

Psychiatric genetics very clearly benefited greatly from rigorous formal genetics theory that ran ahead of (sometimes decades) the empirical science.
What can we learn and generalise?
This piece so far has sung the praises of psychiatric genetics, though if you read closely, the theory that scaffolds the field and its successes is genetic theory, not psychiatric theory. Type-1 error, HARKing and fishing for results are firmly under control because of out understanding of the genome, or arise from social and analytical convention and collaboration that was specific to GWAS. However, any modern GWAS paper breezes past the actual GWAS, and goes on to perform biological annotation, (causal) epidemiological analysis of the psychiatric outcome studied (known as mendelian randomisation), or prediction of case/control status or extern traits based on polygenic scores. These follow up analysis cannot themselves lean on the scaffold afforded by genetic theory, and open the door for selective reporting and HARKing (more likely to report or emphasise success then failures etc). To a degree the strict field specific norms around multiple testingror and GWAS transfer over to those follow up analysis, psychiatric geneticist generally remain fairly vigilant about multiple testing. However, like any science, psychiatric genetics has an imperfect track record when it comes sticking to scientific best practices beyond GWAS. In fact subjectively as a field we may have been slow to pick up on lessons from the broader open-science community, perhaps out of false sense of security based on initial success in GWAS (feel free to view this observations as personal, perhaps others are better at implementing latest best practice than me). Fortunately, a need to keep our practice current haven’t escaped the notice of some in psychiatric genetics. Reseaching this post I learned are primers on how to formally integrate open science practices into all aspects of psychiatric genetics14.
The wide sharing of results, and firm theoretical grounding of GWAS, have enabled identification of a host of biases in our science, a genetic score based on schizophrenia risk predicts dropout/loss to follow up15 , potentially biasing results. GWAS has been used to identify widespread participation bias in modern cohort studies16, GWAS participation in follow up to a primary study found it to be heritable and correlated to all kinds of primary outcomes (biasing analyses)17, and participation bias is sex specific in a way that might mask genuine sex difference18. Access to data, and consistent sharing of results have enabled critical re-evaluation of the (sometimes superficial) measurement of psychiatric outcomes in GWAS19. GWAS has a known and very persistent diversity problem, almost all study particiants arw white, and only recently became somewhat more diverse20. Due to the fundemental soundness, and availability of results and/or data, these biases could readily be studied, papers could be questioned and findings critiqued. However, transparent and well exectuted science does not ensure unbiased science it just enables us to learn about the biases. Good practice does not guarantee genetic findings will translate to biological insight or cure, just like good randomized trials do not guarantee the therapy will work.
Translation in psychiatric genetics will require we take sampling theory, measurement theory, and especially psychological and psychiatric theory as seriously as we take genetic theory. These aren't novel ideas, right before the era of GWAS success Ken Kendler warned that genetics would not solve existing problems in psychiatric nosology21, while Jonathan Flint and Marcus Munafo warned in 207 linking biology to psychiatry via genetics would not be as easy as some made it out to be22. The key ingredients for the success of psychiatric genetics seems to arise from relatively good access to funding, tight type-1 error control, pre-registration (albeit often informally and internally), a social norm that reduces the risk for fishing/selective analysis, systematic measurement
and crucially solid genetic theory.
Lencz, T. et al. (2007). Converging evidence for a pseudoautosomal cytokine receptor gene locus in schizophrenia. Molecular psychiatry, 12(6), 572-580.
Sullivan, P. F. et al. (2008). Genomewide association for schizophrenia in the CATIE study: results of stage 1. Molecular psychiatry, 13(6), 570-584.
Ripke, S. et al. (2013). Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nature genetics, 45(10), 1150-1159.
Singh, T. et al. (2022). Rare coding variants in ten genes confer substantial risk for schizophrenia. Nature, 604(7906), 509-516.
Smith, G. D., & Ebrahim, S. (2005). What can mendelian randomisation tell us about modifiable behavioural and environmental exposures?. Bmj, 330(7499), 1076-1079.
Gottesman, I. I., & Shields, J. (1972). A polygenic theory of schizophrenia. International Journal of Mental Health, 1(1-2), 107-115.
Risch, N., & Merikangas, K. (1996). The future of genetic studies of complex human diseases. Science, 273(5281), 1516-1517.
Pritchard, J. K., & Przeworski, M. (2001). Linkage disequilibrium in humans: models and data. The American Journal of Human Genetics, 69(1), 1-14.
Open Science Collaboration. (2015). Estimating the reproducibility of psychological science. Science, 349(6251), aac4716.
Bulik-Sullivan, B. K. et al. (2015). LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nature genetics, 47(3), 291-295.
a) Smith, D. J. et al. (2016). Genome-wide analysis of over 106 000 individuals identifies 9 neuroticism-associated loci. Molecular psychiatry, 21(6), 749-757.
b ) Okbay, A. et al. (2016). Genetic variants associated with subjective well-being, depressive symptoms, and neuroticism identified through genome-wide analyses. Nature genetics, 48(6), 624-633.
International Schizophrenia Consortium (2009). Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature, 460(7256), 748-752.
Zuk, O. et al. (2014). Searching for missing heritability: designing rare variant association studies. Proceedings of the National Academy of Sciences, 111(4), E455-E464.
Kępińska, A. P., Johnson, J. S., & Huckins, L. M. (2022). Open science practices in psychiatric genetics: a primer.
Martin, J. et al. (2016). Association of genetic risk for schizophrenia with nonparticipation over time in a population-based cohort study. American journal of epidemiology, 183(12), 1149-1158.
van Alten, S et al. (2022). Reweighting the UK Biobank to reflect its underlying sampling population substantially reduces pervasive selection bias due to volunteering. medRxiv, 2022-05.
Adams, M. J. et al. (2020). Factors associated with sharing e-mail information and mental health survey participation in large population cohorts. International journal of epidemiology, 49(2), 410-421.
Pirastu, N. et al. (2021). Genetic analyses identify widespread sex-differential participation bias. Nature Genetics, 53(5), 663-671.
Cai, N. et al. (2020). Minimal phenotyping yields genome-wide association signals of low specificity for major depression. Nature Genetics, 52(4), 437-447.
Mills, M. C., & Rahal, C. (2020). The GWAS Diversity Monitor tracks diversity by disease in real time. Nature genetics, 52(3), 242-243.
Kendler, K. S. (2006). Reflections on the relationship between psychiatric genetics and psychiatric nosology. American Journal of Psychiatry, 163(7), 1138-1146.
Flint, J., & Munafò, M. R. (2007). The endophenotype concept in psychiatric genetics. Psychological medicine, 37(2), 163-180.
Great post thank you!
What do you say to Gwern’s counterpoint that maybe all the culture didn’t amount to much, what mattered was a massive decrease in the cost of genotyping, which unlocked troves of true effects? I.e. there was less reason to hack because truth was hanging low from the tree.
I don’t think it’s the whole story but maybe an important part.